How are biosimilars approved?
Biosimilars undergo an abbreviated development requirement pathway1-3
Fresenius Kabi understands the importance of biosimilars and is committed to the effective and safe treatment of patients. For biosimilar products to be approved, they are required by the FDA and the EMA to pass a series of stringent tests. Specific similarities to the reference product must be demonstrated in the following tests:
Analytical studies: proving that Fresenius Kabi biosimilar products have similar molecular and functional characteristics to the approved product
Clinical studies: clearly illustrating efficacy, safety, pharmacokinetics and pharmacodynamics comparable to the approved drug in the most suitable target patient population and indication
Biosimilar development1-3
Demonstrate molecule is highly similar to the reference product
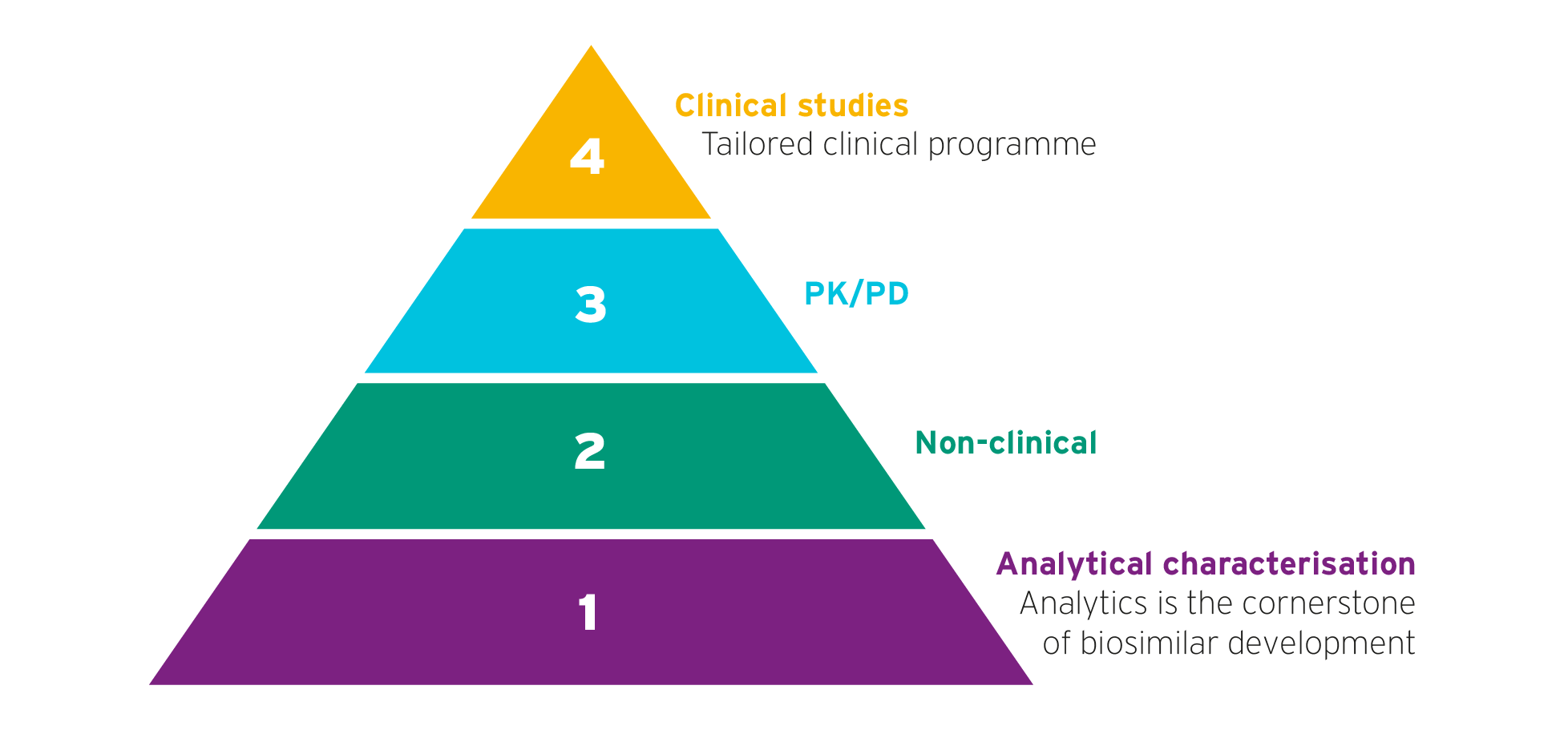
For biosimilars, the emphasis is on the initial stages of development, with a more tailored clinical study program.
Biosimilars undergo an abbreviated development requirement pathway. If a biosimilar is demonstrated to be biosimilar to the reference product, then it is scientifically justified to rely on certain existing knowledge about the safety and efficacy of the reference product to support approval.3
The abbreviated development requirement pathway allows for a potentially more rapid and less costly drug development program for a biosimilar.3
“The evidence acquired over 10 years of clinical experience shows that biosimilars approved through EMA can be used as safely and effectively in all their approved indications as other biological medicines.”1 – EMA
“All FDA-approved biological products, including reference products and biosimilar products, undergo a rigorous evaluation so that patients can be assured of the efficacy, safety and quality of these products.”3 – FDA
EMA
The EU approved its first biosimilar medicine in 2006. In the EU, biologics must be approved through the EMA via their ‘centralized procedure’.1 The review by the EMA/CHMP leads to a ‘scientific opinion’ which is then verified by the European Commission, which can ultimately grant an EU-wide marketing authorisation.1
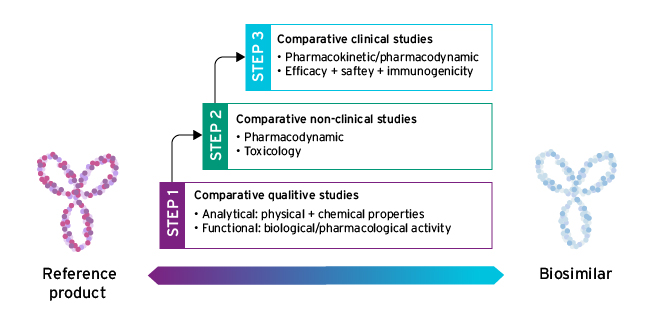
Under the EMA approval process, biosimilar development relies substantially on ‘comparability studies’ to establish biosimilarity to the reference product. This involves a head-to-head comparison of the biosimilar and the reference product.1 This is done via a stepwise process that is tailored to each product: knowledge from the initial quality comparability studies (step 1) is used to identify the extent and type of non-clinical (step 2) and clinical studies (step 3) required in the next step of development, with the aim of ruling out differences in performance between the proposed biosimilar and the reference product.1
The three key steps underpinning biosimilar development in the EU1
US FDA
Through the Biologics Price Competition and Innovation Act (BPCI) of 2009, the US Congress created an abbreviated licensure pathway for biologics that are demonstrated to be biosimilar to or interchangeable with an already FDA-approved biologic.4 This abbreviated licensure pathway was created as a way to provide more treatment options to patients, increasing access to lifesaving medications and potentially lowering healthcare costs.4
An application for the approval of a biosimilar or interchangeable product must include data demonstrating biosimilarity to the reference product. This usually includes data from:
- Analytical studies: demonstrating that the biologic is highly similar to the reference product, in spite of minor differences in clinically inactive components3
- Animal studies, including an assessment of toxicity3
- A clinical study (or studies) that demonstrates safety, purity and potency of the proposed biosimilar product in one or more indications for which the reference product is licensed. This usually includes assessing immunogenicity, pharmacokinetics, and, in some cases, pharmacodynamics, and may also include a comparative clinical study3
In addition to the above, an application for an interchangeable product must also include information or data demonstrating that:
- The proposed interchangeable product is expected to produce the same clinical result as the reference product in any patient3
- For a product administered more than once to a patient, switching between the proposed interchangeable product and the reference product does not lead to increased safety risks or decreased effectiveness compared to using the reference product without any switching between products3
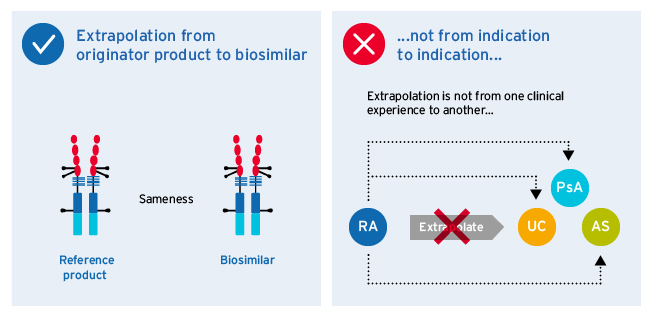
The concept of extrapolation5-7
Extrapolation refers to the approval of a biosimilar for an indication which it has not been evaluated for in clinical trials, based on prior clinical evaluation and approval of the reference product for that indication.5–7
- Analytical comparability confirms that the products are highly similar in terms of physico-chemical properties
- The products are then given to a certain number of patients to verify that they demonstrate highly similar clinical behaviour
- Once this has been demonstrated, the highly similar product can be rolled out to other patients for this indication
Extrapolation enables the development of a biosimilar with several indications through a reduced number of clinical trials,5 thus speeding up the development timeline and giving patients access to more treatment options.
Extrapolation of indications for biosimilars5-7
Why choose biosimilars now and in the future? Learn here
References and footnotes
EMA, European Medicines Agency; FDA, Food and Drug Administration
1. European Medicines Agency (EMA). Biosimilars in the EU: Information guide for healthcare professionals. 2017. Available at:
http://www.ema.europa.eu/docs/en_GB/document_library/Leaflet/2017/05/WC500226648.pdf (Accessed February 2019). 2. McCamish M, Woollett G. Clin Pharmacol Ther 2012; 91:405–417. 3. US Food and Drug Administration (FDA) – Biosimilars. 2018. Available at: https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeutic
biologicapplications/biosimilars/default.htm (Accessed February 2019). 4. US Food and Drug Administration. Biosimilars. 2018. Available at: https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeutic
biologicapplications/biosimilars/default.htm (Accessed February 2019). 5. Mellstedt H et al. Ann Oncol 2008; 19:411–419. 6. Weise M et al. Blood 2012; 120:5111–5117. 7. European Medicines Agency (EMA). Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues (EMA/CHMP/BMWP/403543/2010). 2012. Available at: http://www.ema.Europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf (Accessed February 2019).
